CHMP Meeting Highlights August 2021
Im schriftlichen Verfahren des CHMP im August gibt es keine wichtigen Zulassungsentscheidungen, über die wir in anderen Monaten in den Highlights berichten würden. In dieser Sonderausgabe möchten wir jedoch den 10. Jahrestag der europäischen Zulassung von Ipilimumab würdigen. Diese Zulassung des ersten Checkpoint-Inhibitors (Kontrollpunkte-Inhibitoren) markiert 10 Jahre einer beispiellosen, bis heute anhaltenden Erfolgsgeschichte der Krebsimmuntherapie.
Zehn Jahre Marktzulassung von Ipilimumab, zehn Jahre erfolgreiche Krebsimmuntherapie
1. Erste erfolgreiche Krebsimmuntherapien (1998-2005)
Jahrzehntelang wurde in der Grundlagenforschung und in klinischen Studien versucht, eine sinnvolle Krebsimmuntherapie zu entwickeln. Immer wieder hatten vielversprechende Ansätze letztlich nicht den erhofften herausragenden klinischen Nutzen gebracht. Vor zehn Jahren, im Jahr 2011, sah es trotz großer Hoffnungen so aus, als ob es wieder einmal doch nicht zu einem echten Durchbruch mit neuen Therapieansätzen und einer darauf folgenden Flut innovativer Arzneimittel kommen würde.
Monoklonale Antikörper (mAbs) hatten bereits Einzug in die Krebstherapie gehalten. 1998 erfolgte die Zulassung für Rituximab für die Behandlung von Patienten und Patientinnen mit follikulärem Lymphom in fortgeschrittenen Stadien. Dies belegte, dass die Behandlung von hämatologischen Neoplasien mit mAbs gegen die Oberflächenantigene von Blutzellen ein klinisch sinnvoller Ansatz ist. Zwei Jahre später, im Jahr 2000, zeigte Trastuzumab, ein mAb gegen HER2 auf Brustkrebszellen, dass mAbs gegen Antigene der Zellmembran auch solider Tumore eine beeindruckende klinische Wirksamkeit haben können. Dies wurde bestätigt, als Cetuximab, ein mAb gegen den EGF-Rezeptor, im Jahr 2004 für die Behandlung von Darmkrebs zugelassen wurde. Im Jahr 2005 wurde Bevacizumab zugelassen, das erste Produkt, das erfolgreich den VEGF-VEGF-Rezeptor-Signalweg als pharmakologisches Ziel nutzte.
{kind=link}
Ungeachtet der großen Hoffnungen, dass auf diese bahnbrechenden Produkte bald viele weitere mAbs für die Krebstherapie folgen würden, geschah dies jedoch mehrere Jahre lang nicht. In den Jahren zwischen 2005 und 2011 wurde kein weiterer Wachstumsfaktor oder Wachstumsfaktorrezeptor erfolgreich als Ziel für monoklonale Antikörper in der Krebstherapie genützt, und keine weiteren Zelloberflächenantigene auf Blutzellen konnten als zusätzliche Ziele für hämatologische Neoplasmen etabliert werden.
2. Immunmodulatoren für die Krebstherapie (2005-2011)
Mehrere Jahre lang erhielt kein mAb gegen ein anderes Zielmolekül eine Zulassung für die Behandlung von Krebs. In dieser von 2005 bis 2011 dauernden Lag-Phase der mAbs zur Krebsbekämpfung kam nur eine gemischte Gruppe von "Immunmodulatoren" zur Zulassung. Dabei war die wichtigste Neuerung der Einsatz der Thalidomid-Analoga (2007) zur Behandlung des Multiplen Myeloms (MM) und verwandter Neoplasien. Das Verständnis der genauen pharmakologischen Mechanismen oder der Wirkungsweise von Thalidomid, Lenalidomid und Pomalidomid in der Behandlung des MM ist noch immer unklar, obwohl allgemein angenommen wird, dass eine Stimulierung der Immunreaktion zur Wirksamkeit zumindest beiträgt. Dabei ist auch daran zu erinnern, dass Thalidomid ursprünglich keineswegs als Immuntherapie entwickelt wurde. Die Entdeckung der Wirksamkeit von Thalidomid gegen MM ist eher ein glücklicher Zufall als der Erfolg strategisch geplanter Forschung. Damit ist die Entwicklung von Thalidomid und seiner Analoga zu zulassungsfähigen Produkten auch ein gutes Beispiel für den Wert des „Repurposing“ bekannter Wirkstoffe. Gleichzeitig zeigt es auch Repurposing-spezifische Besonderheiten, wie z. B. die Notwendigkeit einer angemessenen Beteiligung von Opfern der früheren Verwendung einer Substanz am Bewertungs– und Zulassungsprozess. Bei Thalidomid und verwandten Produkten war es wichtig, sowohl die Bedenken der Opfer der früheren Verwendung als auch die Hoffnungen der MM-Patient*innen auf eine künftige Verwendung zu berücksichtigen. Während der Prüfung des Zulassungsantrags für Lenalidomid (das erste in Europa zugelassene Thalidomid-Analogon) lud die Europäische Arzneimittel Agentur (European Medicines Agency, EMA) Thalidomid-Opfer und Myelom-Patient*innen ein, Nutzen und Risiken, einschließlich ethischer Bedenken, zu diskutieren und sich bei der Erstellung des Risikomanagementplans, der Packungsbeilage und der Etikettierung einzubringen.
Im Jahr 2008 erhielt Histamin eine europäische Zulassung für die Behandlung der akuten myeloischen Leukämie (AML) in Kombination mit Interleukin-2. Der pharmakologische Mechanismus ist noch nicht vollständig geklärt, es wird jedoch vermutet, dass er die Immunzellen durch die Hemmung der Bildung reaktiver Sauerstoffspezies schützt.
Im Jahr 2009 wurde Mifamurtid für die Behandlung von Osteosarkomen zugelassen. Mifamurtid ist ein Bestandteil der Zellwand von Mycobakterien und aktiviert Monozyten und Makrophagen zur Sekretion von Zytokinen. Dies erinnert an die frühen Ansätze zur Behandlung von Sarkomen durch bakterielle Toxine Ende des 19. Jahrhunderts. Ungeachtet dieser scheinbar grundlegenden Stimulierung des Immunsystems blieb Mifamurtid jedoch ein Nischenprodukt für die Behandlung von Sarkomen.
Insgesamt schien keines dieser Produkte, die weder wie Rituximab gegen Zelloberflächenantigene von Blutzellen noch wie Trastuzumab oder Cetuximab gegen Rezeptoren von Wachstumsfaktoren gerichtet waren, einen pharmakologischen Ansatz einzuführen, der für eine breite Ausweitung auf andere Tumorarten geeignet schien.
3. Der Durchbruch der mAbs in der Krebsimmuntherapie (ab 2011)
Die Zulassung von Ipilimumab im Jahr 2011 war der Beginn von zehn sehr erfolgreichen Jahren für mAbs und die Immuntherapie von Krebs, und es wurden mehrere neue Zielmoleküle identifiziert und nutzbringend eingesetzt. Wir können drei Hauptgruppen von Zielen für mAbs, die in der Immunonkologie eingesetzt werden, unterscheiden: Zelloberflächenantigene von Blutzellen, die Signalkette von Wachstumsfaktoren und ihrer Rezeptoren und Kontrollpunkte der Immunreaktion (immune checkpoints).
3.1. Zelloberflächenantigene von Blutzellen als pharmakologische Ziele
Seit 2011 sind eine Reihe von mAbs gegen Blutzelloberflächenantigene für die Behandlung hämatologischer Neoplasien zugelassen worden (Abbildung 2). Fast alle Zielantigene sind charakteristisch für die B-Zell-Linie, mit Ausnahme von CD194, das auf T-Zellen exprimiert wird, und CD123, das auf pluripotenten Vorläuferzellen, basophilen Granulozyten und dendritischen Zellen exprimiert wird. Neben "normalen" mAbs wurden auch Antikörper-Wirkstoff-Konjugate gegen CD22 (Inotuzumab Ozogamicin und Moxetumomab Pasudotox), CD79b (Polatuzumab Vedotin) und CD269 (Belantamab Mafodotin) entwickelt. Letzteres ist auch das Ziel der CAR-T-Zellen (Idecaptagene Vicleucel). CD19 ist nicht nur das molekulare Ziel für den "normalen" mAb (Tafasitamab), sondern auch für den bispezifischen Blinatumomab und für die drei CAR-T-Zellen Produkte Axicabtagene Ciloleucel, Tisagene Ciloleucel und KTE-X19.
{kind=link}
3.2. Wachstumsfaktoren und deren Rezeptoren als pharmakologische Ziele
Im Jahr 2000 zeigte die Zulassung von Trastuzumab, dass mAbs gegen Wachstumsfaktorrezeptoren auf soliden Tumoren eine ausgezeichnete Wirksamkeit haben können. Cetuximab, das im Jahr 2004 zugelassen wurde, bestätigte diesen Ansatz. Nach dem Erfolg von Trastuzumab wurden weitere HER2-mAbs, einschließlich konjugierter Antikörper, eingeführt. In ähnlicher Weise folgten auf Cetuximab weitere mAbs gegen EGFR (Abbildung 3). Bemerkenswert ist hier, dass Panitumumab zwar wie Cetuximab gegen EGFR gerichtet ist, aber anders als Cetuximab die Indikation von Panitumumab nicht auf EGFR-exprimierende Tumore beschränkt ist. Necitumumab, das ähnlich wie Cetuximab nur für EGFR-exprimierende Tumore indiziert war, ist inzwischen in der EU nicht mehr erhältlich. Der Zulassungsinhaber (Eli Lilly) von Necitumumab (Portrazza®) beantragte keine Erneuerung der Zulassung, da in der EU keine ausreichende Nachfrage nach dem Produkt bestehe.
In den folgenden Jahren wurden weitere Wachstumsfaktorrezeptoren erfolgreich als Zielmoleküle für die Behandlung solider Tumore eingesetzt (allerdings häufiger mit niedermolekularen Proteinkinase-Inhibitoren als mit mAbs). Die Signalübertragung durch VEGF / VEGFR wurde erstmals als pharmakologisches Prinzip von Bevacizumab (Anti-VEGF) genutzt, das 2005 zugelassen wurde. Später, im Jahr 2014, wurde Ramucirumab zugelassen, das nicht an VEGF, sondern an den VEGF-Rezeptor bindet, wenngleich es nicht das erste Medikament ist, das auf VEGFR abzielt. Vor Ramucirumab waren bereits mehrere kleine Moleküle zugelassen worden, die die Tyrosinkinase des VEGF-Rezeptors hemmen. Gegenwärtig wird die Entwicklung neuer therapeutischer Ansätze, die Wachstumsfaktorrezeptoren als Zielmoleküle nutzen, hauptsächlich von niedermolekularen Proteinkinase-Inhibitoren vorangetrieben. Die Rezeptoren, auf die diese neuen Klassen von Proteinkinase-Inhibitoren abzielen, sind in Abbildung 3 dargestellt.
{kind=link}
3.3. Immunologische Kontrollpunkte als pharmakologische Ziele
Die gegenwärtig erfolgreichste Entwicklung in der Krebsimmuntherapie zeigt die Gruppe der Checkpoint-Inhibitoren. Ipilimumab war der erste Checkpoint-Inhibitor, der jemals für die Behandlung von Krebs zugelassen wurde, hatte jedoch ein begrenztes Wirkungsspektrum. Als Monotherapie ist er nur zur Behandlung des Melanoms zugelassen, zum Einsatz gegen andere Krebsarten muss er mit anderen Checkpoint-Inhibitoren kombiniert werden.
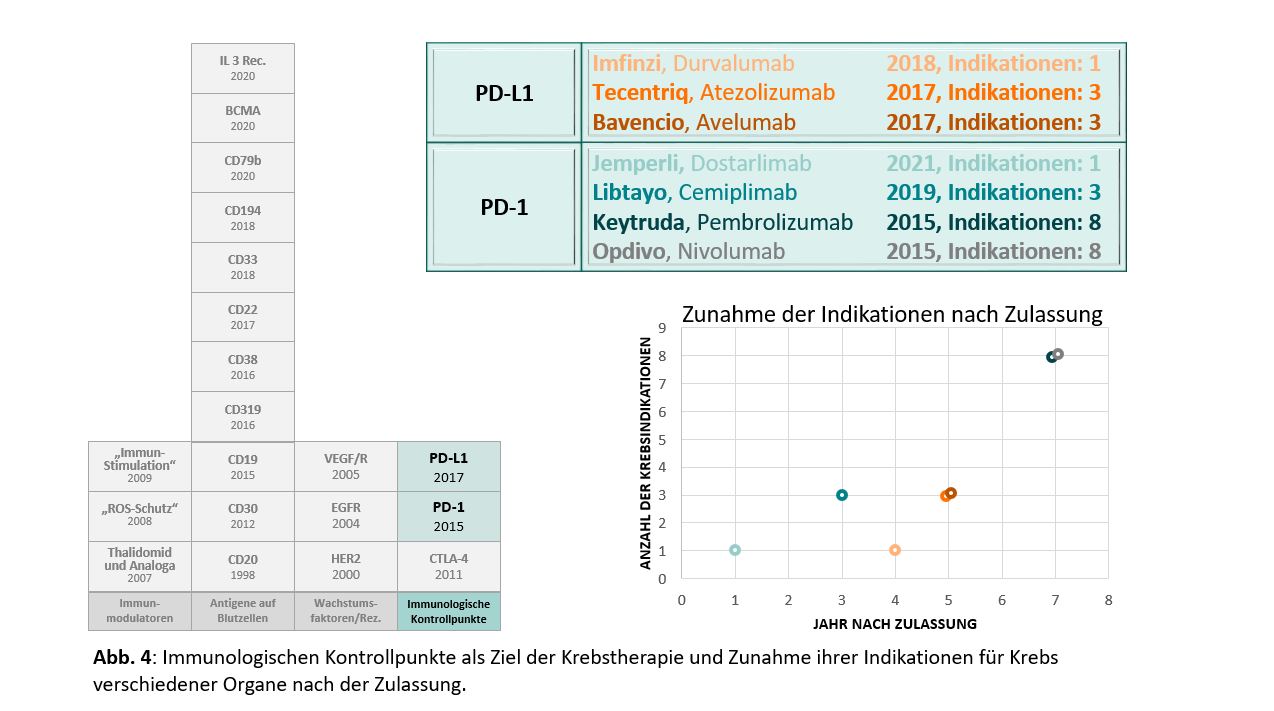
Die pharmakologische Immuntherapie von Krebs wird heute von Substanzen dominiert, die die Interaktion von PD-1 und PD-L1 hemmen und die daraus resultierende Unterdrückung von T-Zellen aufheben. Die Zahl der mAbs, die auf PD-1/PD-L1 abzielen, verdeutlicht die schnelle Entwicklung auf diesem Gebiet. Derzeit, im August 2021, sind sieben mAbs zugelassen (Abbildung 4) und weitere sind in der klinischen Entwicklung oder bereits im Zulassungsverfahren. Gegen kein anderes molekulares Ziel in der Onkologie wurden in so kurzer Zeit so viele mAbs entwickelt. Nur Antikörper gegen CD20 kommen mit drei in der EU zugelassenen Produkten nahe heran, diese wurden jedoch über den langen Zeitraum von 16 Jahren, von Rituximab (1998) bis zu Obinutuzumab (2014) zugelassen.
Dass das Potenzial der Checkpoint-Inhibitoren in der Krebsimmuntherapie bei weitem noch nicht ausgeschöpft ist, zeigt sich auch darin, dass dieselben Produkte immer breiter, d.h. gegen immer mehr verschiedene Krebsarten eingesetzt werden. Während für die altbewährten zytotoxischen und genotoxischen Krebsmedikamente eine Wirksamkeit gegen mehrere Tumorarten üblich ist, haben mAbs für die Behandlung von Krebs in der Regel weniger Indikationen. Nicht nur sind die mAbs gegen Blutzelloberflächenantigene auf hämatologische Indikationen beschränkt, auch mAbs gegen Wachstumsfaktorrezeptoren wie EGFR oder HER2 zielen in der Regel auf Krebs in nur einem oder zwei Organen ab. Breiter gestreute Indikationen finden sich dagegen bei den mAbs gegen VEGF und VEGFR, Bevacizumab (sechs Organe) und Ramucirumab (vier Organe). Dabei ist natürlich zu beachten, dass diese eben nicht direkt gegen die Krebszellen gerichtet sind, sondern die zur Versorgung der wachsenden Tumore notwendige Neubildung von Blutgefäßen als Angriffspunkt haben. Die gegen PD-1 gerichteten Antikörper Nivolumab und Pembrolizumab sind jeweils für die Behandlung von Krebs in acht verschiedenen Organen zugelassen. Diese breite Wirksamkeit weist durchaus Ähnlichkeiten mit der breiten Wirksamkeit der PD-1-Inhibitoren auf, da auch hier eben nicht die Krebszellen, sondern Immunzellen das Ziel der Antikörper sind.
{kind=link}
Die vorherigen CHMP Meeting Highlights sind erreichbar unter: https://www.basg.gv.at/chmp-highlight